
Research
The unified theme of our laboratory is to discover the molecular origins of life - a notion that melds together the multiple focus areas of my tenure home, the School of Molecular Sciences (SMS) and Biodesign Institute (BDI) at Arizona State University (ASU). At BDI’s Center for Applied Structural Discovery, where our laboratory is housed, the design principles of life are sought by determining the structures of biomolecules. Our team of biophysicists, biochemists and computer scientists works in tandem with the center’s structural biologists to discover (i) how phenotypic outcomes (light adaptations, growth and immunogenicity) emerge by tuning the energy transfer between hundreds of proteins embedded in cell membranes or the crowded interiors of cells (Curr. Opin. Struct. Biol 2022, 73, 102338). (ii) Focusing on the ubiquitous family of mitochondrial protein complexes and rotatory F-/V-/A-type ATPases from the AAA-ATPase class of motors, we study their oligomeric architecture and ‘chemomechanical coupling’ as exemplary molecular designs for optimizing biological energy conversion (Frontiers in Physiology 10, 46). (iii) Finally, we determine how leakage in these energy networks lead to ageing, and an uncontrolled increase in the energy turnovers are implicated in health disorders such as, cancer and neurodegenerative diseases (to be summarized in an upcoming invited J. Phys. Chem. B perspective).
Innovations. The primary technique we use to study proteins at the atomistic level is molecular dynamics or MD simulations. In MD, every atom of the system, including the protein, membrane, and explicit water surrounding them, is represented, with the dynamics arising from application of Newton’s Second Law combined with a complex potential function. A grand challenge in simulating chemomechanically coupled dynamics is to concomitantly probe the noisy feedback between fast reactions (10-12 s), slow conformational transitions (10-6–10-3 s), and even slower diffusive processes (10-3 – >1 s). Unique to our program is the development of neural network potential-based flexible fitting (J. Chem. Info. Model. 2020, 60, 2591;J Chem. Phys. 2020, 153, 214102), information theoretic (Matter. 2021, 4, 3195) and geometric machine learning (Nature Comm. 2020, 11, 4734) approaches that allow to visualize cooperativity in molecular ensembles by integrating structural, imaging and kinetic data with MD simulations. We found that learning of continuous transformations from single-particle images offer a typical reaction coordinate for simultaneously tracking chemical reactions, structural transitions and elastic deformations. Dubbed as ManifoldEM, we have applied this method to study the conformational landscape of Calcium pumps, yeast ATPases (Science. Adv. 2020, 6, eabb9605), and spike proteins (Curr. Res. Struct. Biol. 2022, 4, 68). To scale the knowledge of physical features from individual molecules to larger interactomes, we have designed Convolutional Neural Networks (accepted to NeuRIPS, 2022) and Support Vector Machines (Chem 2021, 7, 3393). We bring forth innovations in high performance computing and remote visualization technologies. Our computations scale across thousands of GPU and CPU nodes on the national supercomputers. They concomitantly handle hundred million of interacting particles (J Chem Phys. 2020, 153, 044130), pushing the scope of chemically accurate descriptions to the realm of whole-cell models.
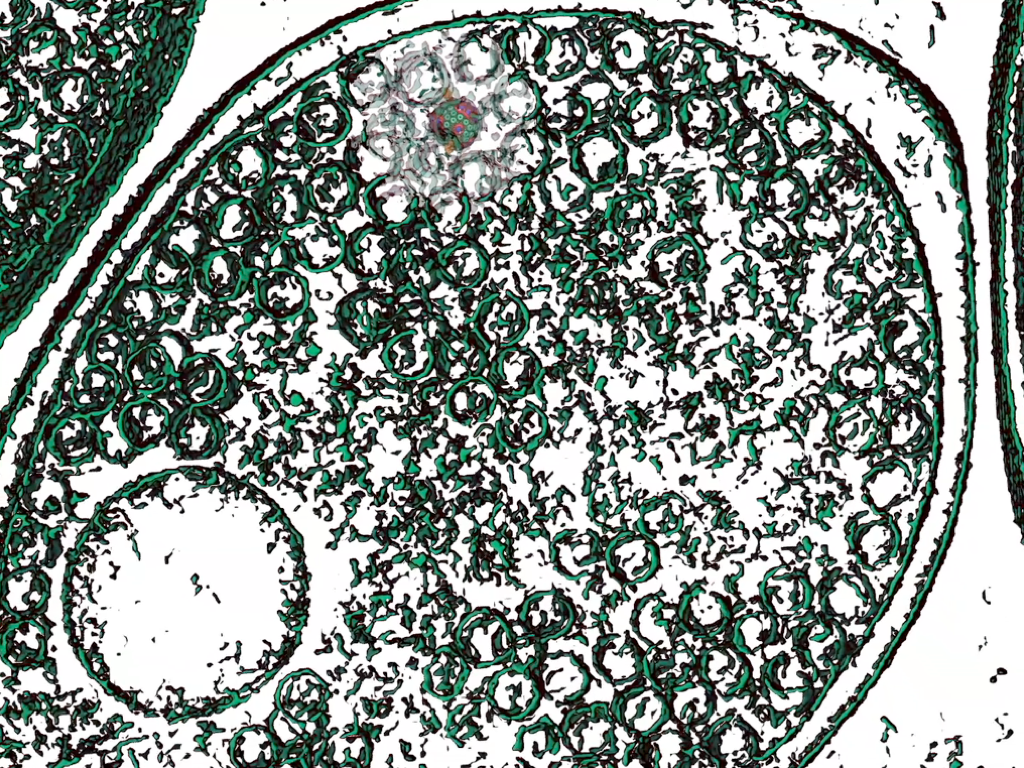
Discoveries and Impact. We have utilized the theory of Brownian Ratchets with molecular simulations to unify the energy storage and dissipation mechanisms across a diverse class of motors (Proc. Nat. Acad. Sci. 2018, 115, 9391). MD simulations have been employed many a time for studying motors. However, many of them have employed external biasing forces or coarse-grained simplifications to derive motor-action. So, it is nontrivial to comprehend whether the directed movements are an outcome of the simulation, or artifacts of the choices made by the simulator. This compromises the generalizability of such method. Instead, we set up a chemical gradient using Monte Carlo methods (BBA Bioenerg. 2020, 1861, 148240) or model chemical changes using free energy perturbation (J. Am. Chem. Soc. 2020, 142, 9220) to induce motor movements within finite-time simulations.
Rectifying a conventional wisdom accumulated over two decades of stationary X-ray structures, we discovered that the energy turnover of oligomeric motors is regulated by tuning not necessarily their structure, rather by controlling the tradeoff between their rate and the reversibility of chemo-mechanical energy transfer (ACS Cent. Sci. 2022, 8, 915). Using so-called string simulations with swarm of trajectories, we found how transitions in macromolecular symmetry can be used as a trick to harness energy storage and optimize utilization. Building on my past work (J. Am. Chem. Soc. 2017, 139, 293), we inferred that the ATPases employ distinct hexameric symmetry-breaking mechanisms for attaining peak rotation speeds vs. maximum thermodynamic reversibility. This creates a competition between the choice of pathways that offer high speed vs. precise clockwise/anti-clockwise rotation. Working with evolutionary biologists, we are now exploring interaction patterns that while conserving ATPases’ structure across organisms from six different clades of life, adjust this rate vs. reversibility competition to adapt with diverse bioenergetic needs. Experimentalists used high-speed AFM to establish that our predicted symmetry-broken forms of the ATPases indeed exist, determined their structures using ATP-inhibitors (independently reviewed in Proc. Nat. Acad. Sci. 120, e2215650120). This also opens the area of controlling reversibility by mutationally tuning its cost of symmetry-breaking. In ongoing work, we are generalizing the idea of symmetry-breaking chemomechanical transitions in the family of dodecamericmeric chemo-sensitive flagellar motors (with A. Shrivastava’s imaging team at ASU) and tetrameric heat-sensitive Transient receptor potential channels (with W. VanHorn’s NMR team at ASU).

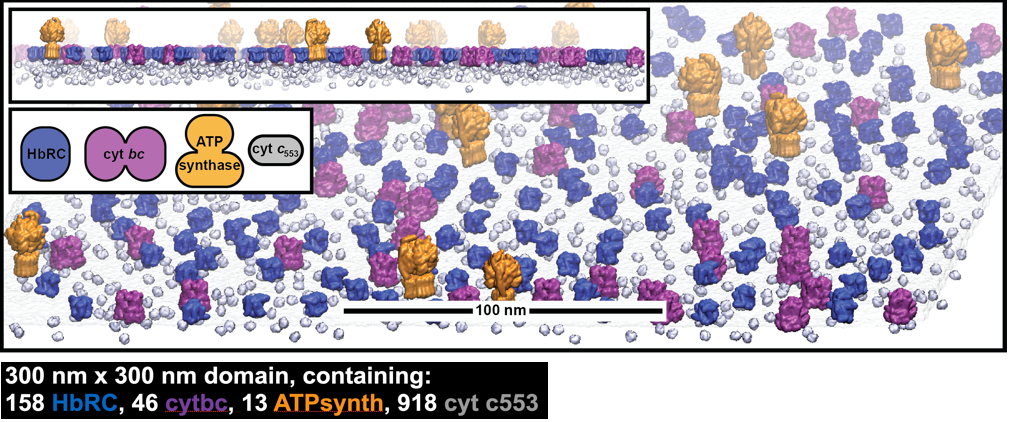
Extending from single-molecule to cell-scale analysis, we performed the first ever MD simulation of an entire organelle (Cell 2019, 179, 1098) to hypothesize that cells control (and sometimes even compromise) the catalytic turnover of individual motors not just to improve the efficiency of network-wide energy transfer but to control their fitness across a range of photo or biochemical stresses. This finding has imminent implications on the molecular evolution of life, and is gaining traction within a broad community of simulators, spectroscopists, chemical and synthetic biologists (see commentary on my work by JD Rochaix). Now, we are extending these fitness models to study the energy metabolism of single-cell organisms (DOE-funded project with synthetic biologist Kevin Redding at ASU), and to study mitochondrial energy transfer in brain cells (with neurobiologist Gulcin Pekkurnaz at UCSD).
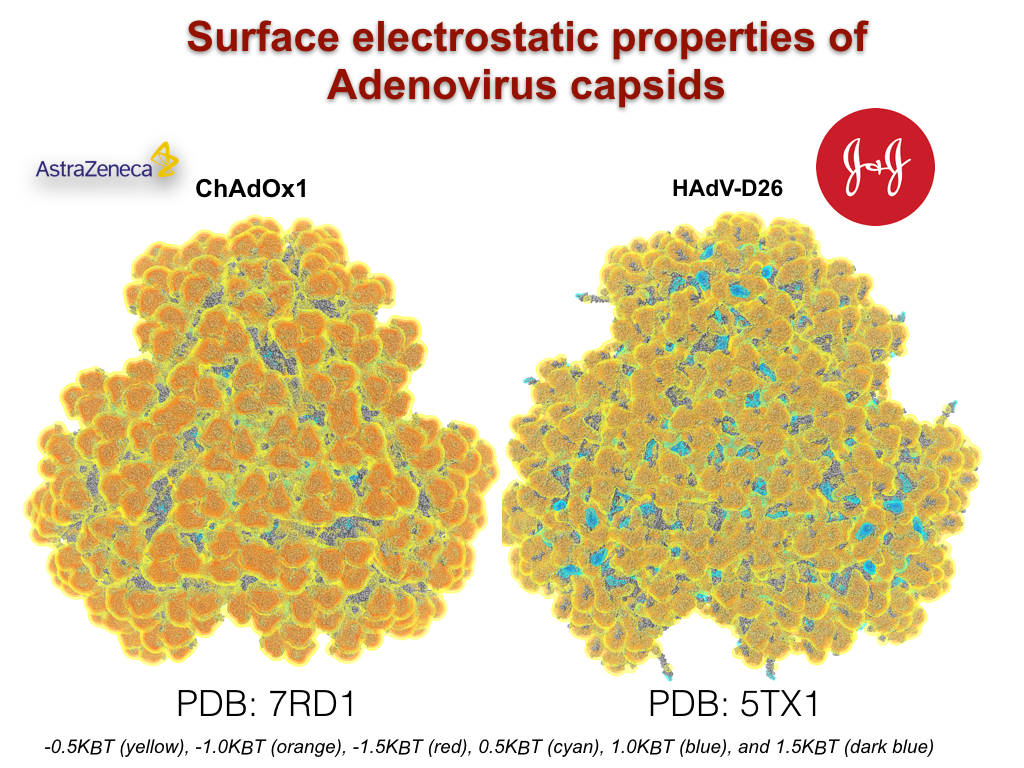
Finally, translating simulations from fundamental biophysics into biomedical applications during COVID19, we determined the molecular mechanism of AstraZenaca and J&J vaccines’s blood clotting by simulating its rare diffusive binding to cytokine proteins (Science. Adv. 2021, 7, eabl8213). We are redesigning AstraZenaca’s vaccine vectors to avoid such therapeutic design failures in the future, which has opened for us new paradigms in personalized medicine and population modeling (Cell Med. 2021, 2, 100221). Together with the Borad team at the Mayo Clinic and a number of colleagues at Mt. Sinai, we are developing bioinformatic tools for predicting patient survival by monitoring the molecular dynamics of their immune-recognition complexes. We have already come up with an immunopeptide prediction method (Cell Systems-in review) that will be leveraged to design T-cell therapies. We have received seed funding through Mayo’s NCI center on developing ‘ex-vivo’ models, and over the next 5 years intend to create an independent program of biophysics-guided personal health between ASU and Mayo Clinic, with NCI and ARPA-H support.
Innovations. The primary technique we use to study proteins at the atomistic level is molecular dynamics or MD simulations. In MD, every atom of the system, including the protein, membrane, and explicit water surrounding them, is represented, with the dynamics arising from application of Newton’s Second Law combined with a complex potential function. A grand challenge in simulating chemomechanically coupled dynamics is to concomitantly probe the noisy feedback between fast reactions (10-12 s), slow conformational transitions (10-6–10-3 s), and even slower diffusive processes (10-3 – >1 s). Unique to our program is the development of neural network potential-based flexible fitting (J. Chem. Info. Model. 2020, 60, 2591;J Chem. Phys. 2020, 153, 214102), information theoretic (Matter. 2021, 4, 3195) and geometric machine learning (Nature Comm. 2020, 11, 4734) approaches that allow to visualize cooperativity in molecular ensembles by integrating structural, imaging and kinetic data with MD simulations. We found that learning of continuous transformations from single-particle images offer a typical reaction coordinate for simultaneously tracking chemical reactions, structural transitions and elastic deformations. Dubbed as ManifoldEM, we have applied this method to study the conformational landscape of Calcium pumps, yeast ATPases (Science. Adv. 2020, 6, eabb9605), and spike proteins (Curr. Res. Struct. Biol. 2022, 4, 68). To scale the knowledge of physical features from individual molecules to larger interactomes, we have designed Convolutional Neural Networks (accepted to NeuRIPS, 2022) and Support Vector Machines (Chem 2021, 7, 3393). We bring forth innovations in high performance computing and remote visualization technologies. Our computations scale across thousands of GPU and CPU nodes on the national supercomputers. They concomitantly handle hundred million of interacting particles (J Chem Phys. 2020, 153, 044130), pushing the scope of chemically accurate descriptions to the realm of whole-cell models.
Discoveries and Impact. We have utilized the theory of Brownian Ratchets with molecular simulations to unify the energy storage and dissipation mechanisms across a diverse class of motors (Proc. Nat. Acad. Sci. 2018, 115, 9391). MD simulations have been employed many a time for studying motors. However, many of them have employed external biasing forces or coarse-grained simplifications to derive motor-action. So, it is nontrivial to comprehend whether the directed movements are an outcome of the simulation, or artifacts of the choices made by the simulator. This compromises the generalizability of such method. Instead, we set up a chemical gradient using Monte Carlo methods (BBA Bioenerg. 2020, 1861, 148240) or model chemical changes using free energy perturbation (J. Am. Chem. Soc. 2020, 142, 9220) to induce motor movements within finite-time simulations.
Rectifying a conventional wisdom accumulated over two decades of stationary X-ray structures, we discovered that the energy turnover of oligomeric motors is regulated by tuning not necessarily their structure, rather by controlling the tradeoff between their rate and the reversibility of chemo-mechanical energy transfer (ACS Cent. Sci. 2022, 8, 915). Using so-called string simulations with swarm of trajectories, we found how transitions in macromolecular symmetry can be used as a trick to harness energy storage and optimize utilization. Building on my past work (J. Am. Chem. Soc. 2017, 139, 293), we inferred that the ATPases employ distinct hexameric symmetry-breaking mechanisms for attaining peak rotation speeds vs. maximum thermodynamic reversibility. This creates a competition between the choice of pathways that offer high speed vs. precise clockwise/anti-clockwise rotation. Working with evolutionary biologists, we are now exploring interaction patterns that while conserving ATPases’ structure across organisms from six different clades of life, adjust this rate vs. reversibility competition to adapt with diverse bioenergetic needs. Experimentalists used high-speed AFM to establish that our predicted symmetry-broken forms of the ATPases indeed exist, determined their structures using ATP-inhibitors (independently reviewed in Proc. Nat. Acad. Sci. 120, e2215650120). This also opens the area of controlling reversibility by mutationally tuning its cost of symmetry-breaking. In ongoing work, we are generalizing the idea of symmetry-breaking chemomechanical transitions in the family of dodecamericmeric chemo-sensitive flagellar motors (with A. Shrivastava’s imaging team at ASU) and tetrameric heat-sensitive Transient receptor potential channels (with W. VanHorn’s NMR team at ASU).
Extending from single-molecule to cell-scale analysis, we performed the first ever MD simulation of an entire organelle (Cell 2019, 179, 1098) to hypothesize that cells control (and sometimes even compromise) the catalytic turnover of individual motors not just to improve the efficiency of network-wide energy transfer but to control their fitness across a range of photo or biochemical stresses. This finding has imminent implications on the molecular evolution of life, and is gaining traction within a broad community of simulators, spectroscopists, chemical and synthetic biologists (see commentary on my work by JD Rochaix). Now, we are extending these fitness models to study the energy metabolism of single-cell organisms (DOE-funded project with synthetic biologist Kevin Redding at ASU), and to study mitochondrial energy transfer in brain cells (with neurobiologist Gulcin Pekkurnaz at UCSD).
Finally, translating simulations from fundamental biophysics into biomedical applications during COVID19, we determined the molecular mechanism of AstraZenaca and J&J vaccines’s blood clotting by simulating its rare diffusive binding to cytokine proteins (Science. Adv. 2021, 7, eabl8213). We are redesigning AstraZenaca’s vaccine vectors to avoid such therapeutic design failures in the future, which has opened for us new paradigms in personalized medicine and population modeling (Cell Med. 2021, 2, 100221). Together with the Borad team at the Mayo Clinic and a number of colleagues at Mt. Sinai, we are developing bioinformatic tools for predicting patient survival by monitoring the molecular dynamics of their immune-recognition complexes. We have already come up with an immunopeptide prediction method (Cell Systems-in review) that will be leveraged to design T-cell therapies. We have received seed funding through Mayo’s NCI center on developing ‘ex-vivo’ models, and over the next 5 years intend to create an independent program of biophysics-guided personal health between ASU and Mayo Clinic, with NCI and ARPA-H support.
Services
As active contributors to the development of the MD simulation software NAMD, our research serves ~20,000 users. We are responsible for the development and dissemination of the molecular dynamics flexible fitting modules inside NAMD, including creation of tutorials and offering workshops. We have offered a number of workshops, including at Harvard Medical School’s SBGRID consortium in 2019, and Stanford Linear Accelerators S2C2 workshop in 2021-2022. Our work is pushing code-automation at multiple national supercomputing centers, where we collaborate with MIDAS team from Brookhaven national laboratory (J. Chem. Info. Model – just accepted). These contributions have put me on the advisory board of NSF’s Jetstream initiative and the Users Executive Board at Oakridge National Lab. I propose software solutions on computational biology to novice and/or non-expert users of national supercomputers. Already being tested in my laboratory, we envision ASU to become the first exascale gateway in the US complementing UIUC that now provide a petascale gateway to all the on-campus researchers. To this end, we have created a remote-visualization website (https://vmd-ood.rc.asu.edu/pun/sys/dashboard) powered by ASU’s Sol supercomputer that allows massively parallel visualization exercises on remote clusters by utilizing simple laptops or tablets with ASU VPN.
Offering due diligence to departmental responsibilities, I serve on the seminar committee for organizing the Departmental seminars on Fridays. I am on the hiring committee for finding a colleague in the area of quantum computing. I am also the departments Social Networking liaison.

My goal as an educator is to create a biophysics pipeline among students of all ages. As a first step, I created the Biophysical Society Student Chapter of the state of Arizona. The chapter, composed of 27 graduate students from ASU and U. Arizona, has established a Biophysics Club in the Arizona College Preparatory school, and is already training 80 students and 17 teachers from 9 more Title 1 schools (3 from Indian reservations) for national and international science competitions. Supported by a seed grant from the Flinn Foundation of Arizona, and subsequently via broader impact activities of NSF and DoD awards, our cloud-based molecular visualization platform serves as a medium for development, distribution and training of K-12 students in computational biophysics. To further the popularity of computers in life sciences, I have organized a BPS Theme meeting on “Biophysics at the Dawn of Exascale Computing” in summer 2022 at Hamburg, and have edited a special volume of Biophysical Journal on the same topic. In addition, I am on the organizing committee of several machine learning conferences including CIKM and Super-Computing conferences. I am cognizant trainee development after graduation from my department. I routinely write recommendation letters for anywhere between ten to twenty students a semester. Our BPS chapter has organized a networking event called Career conversations over coffee in 2019 and 2023 to discuss their career opportunities after grad school. This open-door meeting brought together experts and recruiters from the Intel Corporation, Flinn Foundation, UT Southwestern, Mayo Clinic and Caris Lifesciences. Going beyond just discussions, working with ASU’s the Biotechnology board and local biotech companies I have organized a summer internship program for our students to gain hands-on industrial experience. I am on the advisory board of AZ-based pharmaceutical company i-Metabolomics biopharma, and has just received our first SBIR grant with NIHLB that will further support these internship opportunities.
Offering due diligence to departmental responsibilities, I serve on the seminar committee for organizing the Departmental seminars on Fridays. I am on the hiring committee for finding a colleague in the area of quantum computing. I am also the departments Social Networking liaison.
My goal as an educator is to create a biophysics pipeline among students of all ages. As a first step, I created the Biophysical Society Student Chapter of the state of Arizona. The chapter, composed of 27 graduate students from ASU and U. Arizona, has established a Biophysics Club in the Arizona College Preparatory school, and is already training 80 students and 17 teachers from 9 more Title 1 schools (3 from Indian reservations) for national and international science competitions. Supported by a seed grant from the Flinn Foundation of Arizona, and subsequently via broader impact activities of NSF and DoD awards, our cloud-based molecular visualization platform serves as a medium for development, distribution and training of K-12 students in computational biophysics. To further the popularity of computers in life sciences, I have organized a BPS Theme meeting on “Biophysics at the Dawn of Exascale Computing” in summer 2022 at Hamburg, and have edited a special volume of Biophysical Journal on the same topic. In addition, I am on the organizing committee of several machine learning conferences including CIKM and Super-Computing conferences. I am cognizant trainee development after graduation from my department. I routinely write recommendation letters for anywhere between ten to twenty students a semester. Our BPS chapter has organized a networking event called Career conversations over coffee in 2019 and 2023 to discuss their career opportunities after grad school. This open-door meeting brought together experts and recruiters from the Intel Corporation, Flinn Foundation, UT Southwestern, Mayo Clinic and Caris Lifesciences. Going beyond just discussions, working with ASU’s the Biotechnology board and local biotech companies I have organized a summer internship program for our students to gain hands-on industrial experience. I am on the advisory board of AZ-based pharmaceutical company i-Metabolomics biopharma, and has just received our first SBIR grant with NIHLB that will further support these internship opportunities.